广东泰恩康制药厂有限公司 汕头市 515041
摘要:目的:应用反相高效液相色谱法分离和测定保健食品维生素AD片中维生素A和维生素D3的含量。方法:该供试品经皂化提取和浓缩后,以甲醇定容至10mL,经0.45μm滤过后采用INERTSIL.ODS-SP为色谱柱,甲醇为流动相,流速为1.0mL/min,检测波长分别为325和265nm。结果:维生素A和维生素D3的线性范围分别为2.40~24.0μg/mL和0.8~4.0μg/mL,并与相应的峰面积呈线性关系,相关系数分别为r=0.9990和r =0.9990;测得维生素A和维生素D3的平均回收率分别为98.51%和98.34%。结论:该法可快速、灵敏、准确测定维生素A和维生素D3含量,并应用于日常样品维生素AD片的分析。
关键词:反相高效液相法;维生素A;维生素D3
引言;
维生素A和维生素D3是机体维持正常代谢和机能必需的脂溶性维生素,严重缺乏可致夜盲症、佝偻病等。脂溶性维生素于体内自身合成较难,即便能合成且供应量也不足,需从食物或其他途径中摄取,但该物质又易蓄积于体内造成过量中毒[1] ,故此,对物质应严格控制并定量检测。目前,液相色谱仪法对脂溶性维生素的测定已被广泛的应用。但在实验中发现其维生素A和维生素D3测定的方法过于繁琐、时间长和效率较慢。因此,为了能快速、准确地测定保健食品中的维生素A和维生素D3含量,本文建立其分析方法,并应用于日常样品维生素AD片的分析。
1 仪器与试剂
高效液相色谱仪(日本岛津公司,配备LC-10ATVP型二元溶剂输送泵、SPD-10AVP型紫外-可见检测器、CTO-10ASVP型柱温箱、7725i型手动进样器和2010色谱工作站)、紫外分光光度计(型号TU-1810PC型)、全机械加码分析天平(TG328AS)和电子恒温水浴锅(型号HH·S12·Gr4Ⅱ) 。
维生素A标准品(中国食品药品检定研究院提供,批号100368-201502),标准贮备液(300μg/mL)的制备:精密称取维生素A标准品15mg,加甲醇溶解并定容至50mL棕色量瓶,摇匀,备用。
维生素D3标准品(中国食品药品检定研究院提供,批号100061-201208)标准贮备液(100μg/mL)的制备:精密称取维生素标准品10mg,加甲醇溶解并定容至100mL棕色量瓶,摇匀,备用。
甲醇和环己烷为色谱纯,其它均为分析纯。
2 实验方法
2.1色谱条件
色谱柱为INERTSIL.ODS-SP(250mm×4.6mm,5μm);流动相为甲醇;流速为1.0mL/min,检测波长分别为325nm和265nm[1],柱温为25℃,进样量20μL。
2.2方法[2-3]
2.2.1 样品的制备
碱皂化: 取本品10片,精密称重,研细,精密称取片粉1.0g(约相当于3片量),置于250mL皂化瓶。分别加入蒸馏水5mL、无水乙醇30mL、BHT(2,6-二叔丁基-4-甲基苯酚)0.5g、抗坏血酸1.0g和50%氢氧化钾溶液10mL。再将其置于90℃电子恒温水浴锅进行加热回流45min,时而摇匀,使皂化完全。
萃取:取上述碱皂化液,放冷至室温。将其移入125mL(梨型)分液漏斗,加入环己烷45mL分两次洗涤皂化瓶中残余量,并将洗液一并移入分液漏斗,振摇2min,待环己烷-水相分层后分取水层转移至另一个125mL(梨型)分液漏斗,再加环己烷25mL提取一次,合并环己烷层,以饱和氯化钠溶液洗至中性(以广泛PH试纸检验),加无水硫酸钠5g于预先放置三角漏斗的定性滤纸中。弃饱和氯化钠溶液层,取环己烷层,分液漏斗再用环己烷5mL洗涤,合并环己烷层。
浓缩:将上述环己烷层置于沸点水浴锅下蒸发至约1mL,迅速加入甲醇溶解并定容至10mL棕色量瓶。(若测定维生素A时,则应精密吸取1mL供试液置25mL棕色量瓶,定容至刻度。而测定维生素D3时,则无需再稀释)。经有机系0.45μm滤过后方可直接上机。
2.2.2 测定
按2.1色谱条件项下进行测定,整个过程应避光操作,防止有效成分含量降低。维生素A和维生素D3标准品和供试品色谱图见下图。

维生素A标准品色谱图
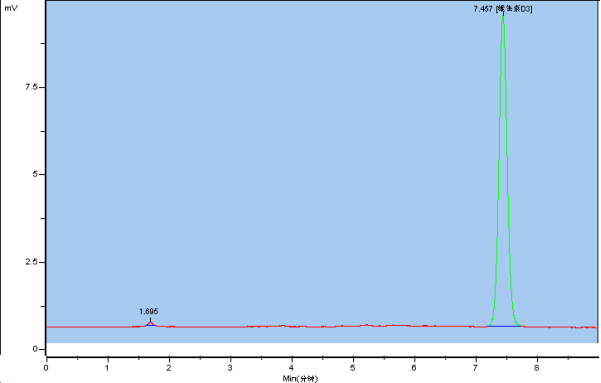
维生素D3标准品色谱图

维生素AD片中维生素A色谱图

维生素AD中维生素D3色谱图
2.2.3 线性范围及检出限
维生素A标准系列浓度工作曲线:精密吸取维生素A标准贮备液0.2mL、0.4mL、0.8mL、1.2mL、1.6mL和2.0mL分别置于不同的25mL棕色量瓶中,加甲醇定容至刻度,摇匀。
维生素D3标准系列浓度工作曲线:精密吸取维生素A标准贮备液0.2mL、0.4mL、0.6mL、0.8mL、1.0mL和1.2mL分别置于不同的25mL棕色量瓶中,加甲醇定容至刻度,摇匀。
按2.1色谱条件项下进行测定,分别以维生素A和维生素D
3的标准系列浓度为横坐标,以各对应峰面积为纵坐标绘制标准曲线,维生素A和维生素D3的线性范围分别为2.40~24.0μg/mL和0.8~4μg/mL,其线性方程分别为Y=3.0501×104X-3.5281×104和Y=1.0946×104X+1.699 ×104,相关系数分别为r=0.9990和r =0.9990。以3倍信噪比计算维生素A和维生素D3的检出限分别为1.0μg/100g和0.5μg/100g。
2.2.4加样回收试验
按2.2项下供试品中的维生素A和维生素D3含量测定方法操作,精密称取已测定供试品含量(维生素A含1.50mg/1g和维生素D3含7.05μg/1g)的样品片粉9份,分别加入不同量标准溶液进行回收率试验,结果见表1和2。
表1供试品中维生素A的加样回收率
组分 | 样品量 g | 本底值 mg | 加入量 mg | 测得值 mg | 回收率 % | 平均回收率 % | RSD % |
维生素A | 1.0088 | 1.51 | 1.20 | 2.72 | 100.83 | 98.51 | 1.27 |
1.0042 | 1.51 | 1.20 | 2.71 | 100.00 | |||
1.0065 | 1.51 | 1.20 | 2.71 | 100.00 | |||
1.0004 | 1.50 | 1.50 | 2.98 | 98.67 | |||
1.0011 | 1.50 | 1.50 | 2.96 | 97.33 | |||
1.0047 | 1.51 | 1.50 | 2.99 | 98.67 | |||
1.0055 | 1.51 | 1.80 | 3.24 | 96.11 | |||
1.0074 | 1.51 | 1.80 | 3.28 | 98.33 | |||
1.0046 | 1.51 | 1.80 | 3.25 | 96.67 |
表2 供试品中维生素D3的加样回收率
组分 | 样品量 g | 本底值/ μg | 加入量 μg | 测得值 μg | 回收率 % | 平均回收率 % | RSD% |
维生素D3 | 1.0088 | 7.11 | 6.00 | 13.02 | 98.50 | 98.34 | 0.99 |
1.0042 | 7.08 | 6.00 | 13.05 | 99.50 | |||
1.0055 | 7.09 | 6.00 | 13.07 | 99.67 | |||
1.0004 | 7.05 | 8.00 | 14.82 | 97.12 | |||
1.0011 | 7.06 | 8.00 | 14.83 | 97.12 | |||
1.0047 | 7.08 | 8.00 | 14.86 | 97.25 | |||
1.0066 | 7.10 | 10.00 | 16.98 | 98.80 | |||
1.0074 | 7.10 | 10.00 | 16.96 | 98.60 | |||
1.0046 | 7.08 | 10.00 | 16.93 | 98.50 |
2.2.5 精密度试验
在同一天内对同一份供试品溶液连续进样6次,以测定值考察其精密度,结果测得维生素A和维生素D3的RSD分别为1.75%和0.92%,实验结果表明该精密度符合要求,见表3。
表3 精密度试验
第1次 | 第2次 | 第3次 | 第4次 | 第5次 | 第6次 | RSD% | |
维生素A/ mg | 1.51 | 1.52 | 1.49 | 1.45 | 1.50 | 1.47 | 1.75 |
维生素D3/μg | 7.20 | 7.15 | 7.09 | 7.03 | 7.05 | 7.06 | 0.92 |
2.2.6供试品分析
按2.2项下测定维生素A D片中的维生素A和维生素D3含量方法操作,共测定6批次,并与标准方法[4]进行了比较,实验结果见表4。
表4 6批次样品含量测定结果方法对比
| 本法 | 标准法 | ||
维生素A/mg | 维生素D3/μg | 维生素A/mg | 维生素D3/μg | |
131001 | 1.51 | 7.22 | 1.48 | 7.19 |
131002 | 1.40 | 7.45 | 1.45 | 7.49 |
131003 | 1.36 | 7.36 | 1.35 | 7.39 |
131004 | 1.47 | 7.48 | 1.42 | 7.43 |
131005 | 1.30 | 7.78 | 1.27 | 7.72 |
131006 | 1.27 | 7.68 | 1.34 | 7.64 |
3 讨论
3.1皂化条件优化[5]
由于维生素A既可以脂肪酯之形式,又可以棕榈酸酯之形式存在,且其理化性质各有差异,而维生素AD片中的维生素D3含量偏低,加上辅料的干扰,均会可影响测定结果,给实验上带来困难。因此,应进行皂化处理以提高其纯化程度。
本文中的皂化处理不仅将供试品中脂肪等成分水解转变成水溶性物质而除去,加之而且其碱性皂化还可以除去样品的其它杂质在测定过程中,皂化条件必须严格控制,若皂化条件太温和(温度≤70℃)会使维生素A 成分水解以致皂化不完全;若皂化条件太猛烈(温度≥90℃)则可使测定结果的重现性差以致试验无意义。
3.2供试品提取溶剂的选择
由于维生素A和维生素D3都是均为脂溶性维生素,故选择环己烷和乙醚有机溶剂作为提取溶剂,按上述方法对其进行处理,实验结果发现两者溶剂均能取得良好的提取效果,但由于乙醚是属于管制易制毒试剂,考虑到使用上的不方便且毒性较大,故最终选择环己烷为提取溶剂。
3.3 系统最佳实验条件选择
3.3.1流速的选择
2010版中国药典规定[4]:维生素A所属峰面积之分离度应>3.0和维生素D3所属峰面积之分离度应>1.0。为了符合上述规定,将其标准溶液进行流速之选择:若流速调至1.5mL/min时,维生素A峰面积之分离度>3.0,而维生素D3峰面积之分离度<1.0;若流速调至1.0mL/min时,维生素A峰面积之分离度仍>3.0,而维生素D3峰面积之分离度>1.0;即是当流速调至1.5mL/min或1.0mL/min时,维生素A峰面积基本一致,而维生素D
3则有明显差别。因此,实验中选择流速为1.0mL/min。
3.3.2测定波长的选择
采用紫外分光光度计,在200~400nm处对浓度为维生素A(3.6μg/mL)和维生素D3(1.2μg/mL)的标准溶液进行波长扫描,以确认其最大吸收峰,实验结果表明,上述维生素A和维生素D3标准溶液的最大吸收波长分别为325nm和265nm处。
4 结论
本文对维生素A D中所含的维生素A和维生素D3进行皂化-萃取-浓缩等步骤处理,并采用液相色谱技术,使维生素A和维生素D3得到良好分离和取得较为满意的测定结果。在测定的同时,与标准方法进行了比较,两者测定结果基本一致,相对误差均符合国家标准方法中的维生素A≤算术平均值的5%和维生素D3≤算术平均值10%的规定要求[6]。该法操作简便、快速 、重现性好、分离度好、大大缩短测定时间;供试品回收率较高,稳定性好,结果准确,可作为日常样品维生素AD片的快速分析。
参考文献
[1]曾红燕,邹晓莉,黎源倩,许见勤.HPLC同时测定保健食品中维生素A、D3、E和β-胡萝卜素[J].分析试验室,2008;27( 2):15-18
[2]黄双路,罗红斌,陈伟. 维生素A胶丸含量测定方法的研究[J].中国药品标准,2003;(04)03:41-42
[3]WMJ-WI-ZG-78,食品中维生素A、D3含量测定操作规程[S]
[4]中国药典(2015版),四部[S]维生素A测定法、维生素D3测定法:通则0721-0722
[5] 裘立群, 高艾英 ,张昊, 井华. 高效液相色谱法测定水果中脂溶性维生素含量的研究[J].广州化工2010;38(10):151-153
[6]GB/T5413.9-2010,食品安全国家标准方法[S]
1

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



