空军军医大学第一附属医院 儿科 陕西 西安 710032
摘要:目的:患儿,男,6岁,因发现肾功能衰竭为首发症状就诊,伴有肥胖、多趾(指)、视力下降就诊,经基因检测确诊为Bardet-Bidel综合征,因此,对于肾功能衰竭患者,需积极寻找肾衰竭的原发病,若合并视力异常、多指(趾)、肥胖等,需警惕BBS可能。通过回顾性分析该患者的诊断和治疗过程,查阅国内外相关研究,旨在提高临床医生提高临床医师对本病的认识。
关键词:肾功能衰竭;BBS;多指(趾)
1.病例资料
患儿,男,6岁,以“发现肝功能异常10天,尿素氮、肌酐升高3天”于2020年08月入院。入院前因入学体检时发现转氨酶高,无腹部不适感,后详细查尿素氮、肌酐升高,无恶心、呕吐,无尿量增多、泡沫尿、血尿,无多饮、多食等症状, 1天前来我院时出现发热,体温为37.8℃,伴有鼻塞,无咳嗽,有呼吸增快。既往史:患儿系“遗弃儿”,血缘父母无法追踪,于2月龄大小时开始较同性别、同龄儿童体型肥胖,3岁时因“双下肢向左偏斜”在器具辅助下行“双下肢矫正术”,平素有夜间视力下降。查体:血压:90/60mmHg,身高:114cm,体重:31Kg,BMI:23.85Kg/m2,(正常同性别、同龄儿P3-P97百分位为13.15-18.6),体型肥胖,嘴角向下,反应稍迟缓,四肢活动稍笨拙,颈部淋巴结可触及数个肿大,最大者直径为2.0×1.0cm,活动度好,无压痛,咽部黏膜充血明显,可见疱疹破溃后改变。双手(足)为六指(趾)。(图1)

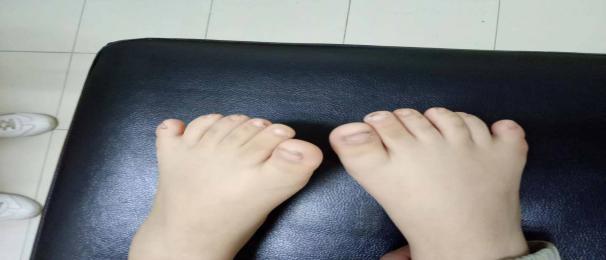

图1 患儿外观 双手(足)为六指(趾)
实验室检查:血常规:血红蛋白:83g/L,平均红细胞体积:85.6fl,平均血红蛋白含量:27.9pg,平均血红蛋白浓度:325g/L,尿常规:尿比重1.007,尿PH值5.5,尿蛋白2+,血气分析:PH7.28,pO2:92mmHg,pCO217mmHg,BE(B):-17.3mmol/L,HCO3-:8.0mmol/L,肝功能:天门冬氨酸氨基转移酶:53IU/L,丙氨酸氨基转移酶:115 IU/L,肾功能:尿素:774umol/L,肌酐:29.30mmol/L,胱抑素C:5.50mg/L。24小时尿蛋白定量:1.11g,肾小球滤过率:9.85 ml/min,内生肌酐清除率:4.6ml/min,双手X线片:双手小指侧多指畸形,双手骨质疏松。双足X线片:双足多趾畸形。腹部B超:左肾大小约5.3×2.8×3.5cm,可见数个无回声,右肾大小约6.6×2.9×3.3cm,可见数个无回声,皮髓质分界不清,双肾小,弥漫性病变,双肾囊肿。
眼科专科:视力:右侧0.08,左侧0.1。眼底:视网膜色淡,色素不均匀。OCT提示:双眼外层视网膜萎缩,椭圆带缺失。EOG:提示视网膜色素上皮功能降低。双眼视觉电生理:双眼F-ERG各项反应幅值重度降低,P-VEP峰时延迟,波形存在,双眼LP、Arden比值均降低,暗适应检查提示暗适应能力下降。
经患儿家属知情同意后抽患儿静脉血2ml,采用凯昂设计的聚合全外显子探针进行文库捕获,在 Illumina 公司 Novaseq 系列测序仪进行文库测序,全外显子检测 (北京凯昂实验室) :发现与受检者临床表型相关的变异:BBS4 基因,c.1104dupT Likely pathogenic(疑似致病性变异),本例中患儿基因检测为,BBS4基因 (NM-033028) 杂合突变, 突变位点为c.1104dupT,p.D368fs, 染色体位置chr15-73027520-73027520,在OMIM数据库(人类孟德尔遗传)编号为615982。相关报道Katsanis 等(2002)对1名Bardet-Biedl综合征患者进行基因检测,在 BBS4 基因的 4 号外显子上检测到一个纯合剪接变异位点,IVS3-2A-G。该变异在正常人群数据库中未见携带。经预测,该变异会导致RNA 剪接异常,为致病性变异。
诊疗经过:入院后诊断为Bardet-Bidel综合征,给予低盐饮食,抗感染、碳酸氢钠纠酸、尿毒清、药用炭片保肾等对症治疗13天后好转出院,随访2月后于外院开始规律腹膜透析治疗。
2.讨论
Bardet-Bidel综合征(BBS)是一种罕见病,为常染色体隐性遗传的疾病,患病率为1/150000-1/140000,存在地区差异[1]。
BBS诊断标准有主要特征和次要特征, 主要特征包含有:视网膜营养不良、多指 (趾) 、肥胖、学习障碍、男性性腺发育异常和肾脏异常;次要特征包括:发育障碍、斜视/白内障/散光、短指/并指、生长发育、肾性多尿多饮、共济失调/平衡能力差、轻度痉挛 (尤其指双下肢) 、糖尿病、牙齿拥挤/缺牙/小牙根/高腭弓、左心室增大/先天性心脏病及肝脏纤维化。满足4个主要特征或满足3个主要特征+2个次要特征即可诊断BBS
[1-2]。主要特征:(1)视网膜变性[3],最常见,占90-100%,早期可出现,主要表现为夜盲、视野缩小、中心视力下降,色觉丧失,出现全盲的平均时间为15.5岁;(2)肥胖[1],占比72-92%,婴儿期可出现,早期全身均匀肥胖,成年后为向心性肥胖;(3)多指 (趾),发生率为59-81%,出生时即可发现,可为单侧、双侧,对称或不对称;(4)智力低下,发生率为50-61%,程度不一,常伴语言表达较同龄儿童迟缓,面容呈痴呆状;(5)生殖系统异常,占比59-98%,男性大多不育,仅有2.7%女性可正常生育;(6)肾功能异常,发生率占90%以上[4-5],引起BBS死亡的主要原因。针对于本例患儿,满足主要4个特征,临床诊断成立,因该患儿家族史情况不明,因此早期未能及时诊断,BBS的许多临床表现是随年龄增长而逐渐出现的,在无家族史的情况下,很少能早期确诊。
BBS发病至少与21个基因突变有关,即BB1-21,80%患者可通过基因检测明确诊断[6-7]。各基因突变在BBS患者发病过程的作用不尽相同, 具有基因异质性,当BBS基因突变引起所编码的蛋白质改变时, 可导致纤毛的组装过程或信号转导复合物的形成受阻, 可以引起相应的疾病, BBS是一种纤毛功能缺陷疾病[8-9],BBS4基因型突变大多在中东或者北非报道[10]。不同基因型,患儿临床表象不一致,基因型与表型有一定相关性。既往报道的BBS4基因肾损害严重,该患儿早期出现肾功能衰竭,与既往报道相符合。该患儿肝功异常,本次考虑为EB病毒感染所致,若后期再次出现反复增高,需警惕肝纤维化可能。
Bardet-Bidel综合征目前没有特殊治疗方法,主要以对症治疗为主[11-12],针对于本例患儿,以保肾、控制饮食、降低体重为主,肾衰竭可能为预后较差的主要原因,有肾衰竭患者, 应积极寻找肾衰竭的原发病, 若有多系统受累, 提示遗传性疾病BBS可能,条件允许者应尽早完善相关基因检测, 产前诊断有助于提高早期明确胎儿是否是携带或患病,尽早诊断、尽早干预, 以改善预后。
参考文献
[1]Beales PL, Elcioglu N, Woolf AS, et al.New criteria for improved diagnosis of Bardet-Biedl syndrome:results of a population survey[J].J Med Genet, 1999, 36 (6) :437-446.
[2]David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, et al. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes[J]. J Med Genet, 1999, 36(8): 599-603.
[3]Andrade LJ,Andrade R,Frana CS,Bittencourt AV. Pigmentary retinopathy due to Bardet-Biedl syndrome: case report and litera- ture review[J].Arq Bras Oftalmol,2009,72( 5) : 694-696.
[4]沈明,林燕,庞宁.Bardet—Biedl综合征的研究进展[J]. 中华儿科杂志,2001,39(12):764 766.
[5]Putoux A,Attie-Bitach T,Martinovic J,Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney[J].Pediatr Nephrol,2011 Jan 19.[Epub ahead of print]
[6]Heon E, Kim G, Qin S, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21) [J].Hum Mol Genet, 2016, 25(11):2283-2294.
[7]M'hamdi O, Ouertani I, Chaabouni-Bouhamed H.Update on the genetics of Bardet-Biedl syndrome [J]. Mol Syndromol, 2014, 5(2):51-56.
[8]Warburg M, Riise R.Bardet-Biedl and Cohen syndromes:differential diagnostic criteria[J].J Med Genet, 2000, 37 (12) : E46.
[9]沈涛,严新民,肖春杰.巴德一毕氏综合征研究的现状及意义 [J].中华医学遗传学杂志,2013,30(5):570—573.
[10]Forsythe E, Beales PL. Bardet-Biedl syndrome [J]. Eur J Hum Genet, 2013, 21(1): 8-13.
[11]Alvarez-Satta M, Castro-Sanchez S, Valverde D. Bardet-biedl syndrome as a chaperonopathy: Dissecting the major role of chaperonin-like bbs proteins (bbs6-bbs10-bbs12). Frontiers in molecular biosciences ,2017,4:55.
[12]成胜权,曹玉红,张劲松,等. Bardet-Bidel综合征1例报告[J].中国当代儿科杂志,2013.11(18):975-976.
通信作者:成胜权

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



